Oluline on alati arvestada lugemispikkust ja kvaliteeti koos suure vigadega lugemisandmetega ning praegustel kaua loetud tehnoloogiatel (nt MinION ja PacBio) on kõrge veamäär. Lugemispikkuse ja kvaliteedi ühine arvestamine aitab teil kindlaks teha, kui edukas oli jooks, mitu lugemist oli "kõrge kvaliteediga", kas pikemad lugemised on "päris" (või lihtsalt poorimüra) jne.
Olen hiljuti sarnaste süžeede vastu huvi tundnud ja kohtasin projekti nimega pauvre (prantsuse keeles "vaesed", mängivad "pooridel" ') Oxford Nanopore Technologies (ONT) kogukonna kaudu, mis on minu arvates isegi parem kui MinKNOWi baaskõne plaan. Lisaks saate need graafikud kiirelt failist genereerida, kui soovite, erinevalt MinKNOW-st.
[Märkus: ma ei ole algne autor, kuid panustan nüüd, sest see meeldis (ja vajas).]
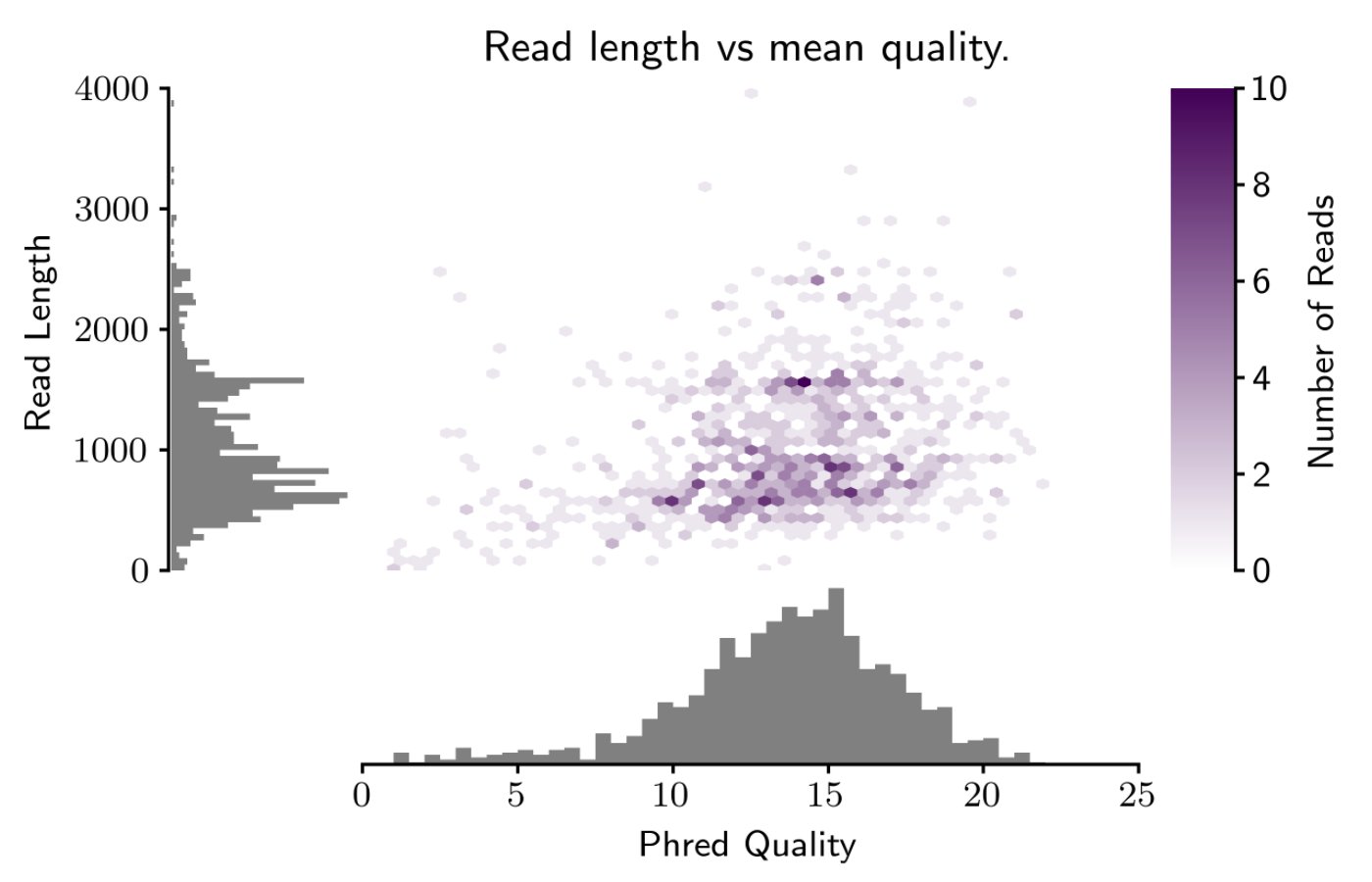
Pauvre teatab ka kasulikust statistikast:
fastq statistika fastq_runid_bb8b8ddedb22bdd6802b2bfa2b4e424c92c30d28_0.fastqnumReads: 2164829numBasepairs: 4970615 : 1495.0minLen: 5maxLen: 392031N50: 3450L50: 402786 Aluspinnad > = prügikast keskmise PHRED ja pikkuse järgi minLen Q0 Q5 Q10 Q15 Q17.5 Q20 Q21.5 Q25 Q25.5 Q30 0 4970615217 4970611559 4835461771 3810997 681104153154153104153104103154103104154003104154004 270324 160128 50729 50729 0 0 0 0100000 6260554 6260554 0 0 0 0 0 0 0 0 0150000 3504240 3504240 0 0 0 0 0 0 0 0200000 2501101 2501101 0 0 0 0 0 0 0 0
250000 1609592 1609592 0 0 0 0 0 0 0 0300000 1033423 1033423 0 0 0 0 0 0 0 0350000 392031 392031 0 0 0 0 0 0 0 0 Lugemiste arv > = bin keskmine Phred + LenminLen Q0 Q5 Q10 Q15 Q17.5 Q20 Q21.5 Q25 Q25.5 Q30 0 2164829 2164605 2083436 1626706 1183812 435687 77341 1 0 0 50000 109 109 5 3 1 1 0 0 0 0100000 36 36 0 0 0 0 0 0 0 0150000 15 15 0 0 0 0 0 0 0 0200000 9 9 0 0 0 0 0 0 0 0250000 5 5 0 0 0 0 0 0 0 0 0300000 3 3 0 0 0 0 0 0 0 0350000 1 1 0 0 0 0 0 0 0 0
Need graafikud ja statistika oleksid PacBio puhul võrdselt kasulikud, kuid see pole super lihtne (kuigi see on võimalik) Sequel sequenceri praeguse tooraine väljundiga: Millist kvaliteediskooride kodeeringut PacBio kasutab?
Pauvre kasutab praegu Biopython parsida tegeliku joonise jaoks fastq ja matplotlib ning lasta valida väljundkujutise vorming (nt .png, .pdf jne). Samuti saate valida, kas taust on läbipaistev või valge (.png-väljundi jaoks).
Parser on praegu üliaeglane, kuna see kasutab SeqIO.parse , kuid selle kiirendamiseks vahetame parsereid. Lisame ka mõned lisafunktsioonid (nt valige, kas lisada Y-teljed veeriste histogrammidesse, printida osa statistikat dokumenteerimiseks otse joonisele)
Lilla on praegu ainus värvivalik (mida ma isiklikult armastan), kuid muutmisvalikute lisamine on ülilihtne.